NGS Basic Analyses
Next-generation sequencing (NGS) technologies is able to accelerate genetics and epigenetics research. For examples, DNA-seq for whole genome assembly, mRNA-seq for transcriptome studies, ChIP-seq for protein-DNA binding and histone marks, ATAC-seq for chromatin accessibility, and BS-seq for DNA methylation profiling (Fig.1).
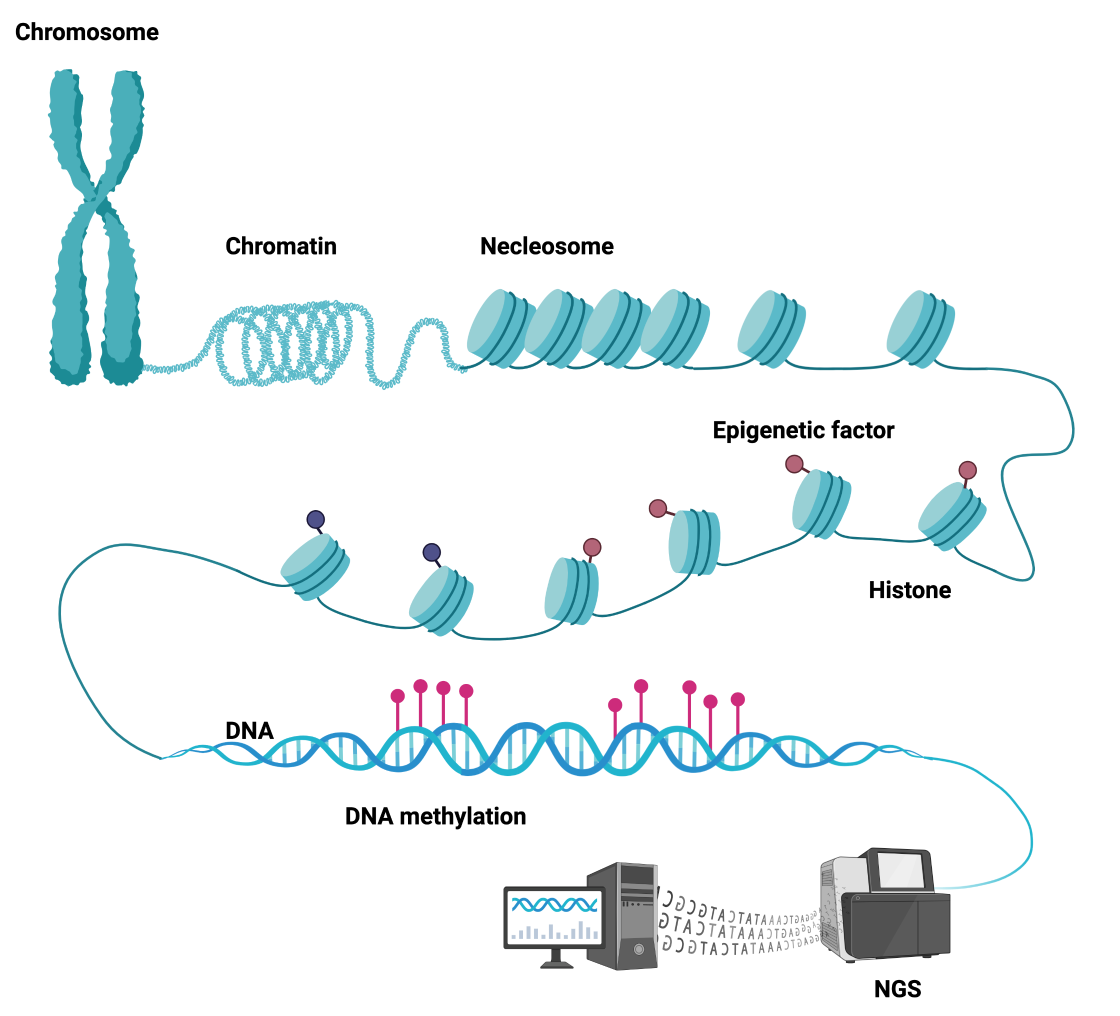
Fig1. Schematic of genetics and epigenetics applying NGS technologies.
Background
Download raw data from NCBI SRA
Sequence Read Archive (SRA) stores raw sequencing data and alignment information for high throughput sequencing ,NGS for instance, data can be download via latest SRA Toolkit1.
prefetch SRR957710
# 2022-12-14T07:23:37 prefetch.2.11.2: Current preference is set to retrieve SRA Normalized Format files with full base quality scores.
# 2022-12-14T07:23:38 prefetch.2.11.2: 1) Downloading 'SRR957710'...
# 2022-12-14T07:23:38 prefetch.2.11.2: SRA Normalized Format file is being retrieved, if this is different from your preference, it may be due to current file availability.
# 2022-12-14T07:23:38 prefetch.2.11.2: Downloading via HTTPS...
# 2022-12-14T07:23:42 prefetch.2.11.2: HTTPS download succeed
# 2022-12-14T07:23:42 prefetch.2.11.2: 'SRR957710' is valid
# 2022-12-14T07:23:42 prefetch.2.11.2: 1) 'SRR957710' was downloaded successfully
The sra data will then be found in the folder ncbi/public/sra/ or current path. To convert the sra to fasta file, another tool in SRA Toolkit can be applied.
fastq-dump SRR957710.sra ## if single-end
fastq-dump --split-files SRR957710.sra ## if paired-end
# Read 42422649 spots for SRR957710.sra
# Written 42422649 spots for SRR957710.sra
Then the raw fasta/fastq file data is able to used in the following NGS analyses.
Check data
When downloading files for patches or drivers, it is important to ensure that the file is complete and hasn't been corrupted during the download process. One way to verify the integrity of the file is by comparing its MD5 checksum.
## create a md5 file of three data for others
md5sum data1.fastq data2.fastq data3.fastq > md5.txt
# c1fa0dfbd869b01995b0818e4d176011 data1.fastq
# 5f6a6c6f121351a1b7f33a2372df9932 data2.fastq
# c170b6cd45b9f2a2ff580c4837e5bb8a data3.fastq
## check md5 after download the data
md5sum -c md5.txt
# data1.fastq: OK
# data2.fastq: OK
# data3.fastq: OK
even the change is so small the md5 checksum will be different
Quality control
It is recommend to check the check the quality for the raw data beforehand.
fastqc SRR957710.fastq
Users can view the result SRR957710.html via web browser.
Trimming data
After the viewing the output of fastqc. The suggested low quality reads and adaptors should be removed by the trimmimg tools such as Trimmomatic.
java -jar /Trimmomatic-0.39/trimmomatic-0.39.jar PE -phred33 ## paired-end
SRR957710_R1.fastq.gz SRR957710_R2.fastq.gz ## input
SRR957710_R1_paired.fastq.gz SRR957710_R1_unpaired.fastq.gz ## output of R1
SRR957710_R2_paired.fastq.gz SRR957710_R2_unpaired.fastq.gz ## output of R2
ILLUMINACLIP:/Trimmomatic-0.39/adapters/NexteraPE-PE.fa:2:30:10:8:true ## remove adaptor
LEADING:3 ## remove low quality
TRAILING:3 ## remove low quality
SLIDINGWINDOW:4:15 ## screening window size
MINLEN:36 ## drop reads below 36 bases long
Alignment
The raw sequences (DNA or RNA) needed to be aligned to their own reference genome by the similarity, identifying the regions of reads.
Where to download the reference genome?
iGenomes is a collection of reference sequences and annotation files for commonly analyzed organisms.
There are several common used alignment tools such as bowtie2 2 can be applied in the analyses. The libraries like RNA-seq or BS-seq are recommend using their own aligners.
bowtie2 -p 30 -x reference_genome -U SRR957710.fastq -S SRR957710.sam
# -p number of thread (default=1)
# -x path to reference genome
# -S output sam file
Mapping table
After alinment, the mapping table is required in most of the analysis so that other can esily understand the status of the libraries and as well as the data.
| Samples | Raw reads | Mapped reads | Maapability (%) |
|---|---|---|---|
| Sample1 |